
II类医疗器械所需申请资料
1) 申请函,此部分应包括申请人(或联系人)和企业的基本信息、510(K)递交的目的、申请上市器械的名称型号和分类资料、进行实质等效比较的产品(Predicate Device)名称及其510(K)号码;
2) 目录,即510(K)文件中所含全部资料的清单(包括附件)
3) 真实性保证声明,对此声明,FDA有一个标准的样本;
4) 器材名称,即产品通用名、FDA分类名、产品贸易名;
5) 注册号码,如企业在递交510(K)时已进行企业注册,则应给出注册信息,若未注册,也予注明;
6) 分类,即产品的分类组、类别、管理号和产品代码;(7) 性能标准,产品所满足的强制性标准或自愿性标准;
8) 产品标识,包括企业包装标识、使用说明书、包装附件、产品标示等;
9) 实质相等性比较(SE);
10) 510(K)摘要或声明;
11) 产品描述,包括产品的预期用途、工作原理、动力来源、零组件、照片、工艺图、装配图、结构示意图等;
12) 产品的安全性与有效性,包括各种设计、测试资料;
13) 常规测试项目: 生物相容性;产品性能。
14) 色素添加剂(如适用);
15) 软件验证(如适用);
16) 灭菌(如适用),包括灭菌方法的描述、灭菌验证产品包装和标识等。
四、向FDA提交510(k)文件进行评审
递交的510(K)文件的格式完整性(即510(K)文件行政性审核);
递交的510(K)文件的安全和有效性论证的符合性(即510(K)技术审核)并尽量避免和减少FDA的审核提问;
递交的510(K)文件的所有产品信息,测试报告等基本符合FDA认可的法规,标准和导则要求,不会有重大疏漏和错误。
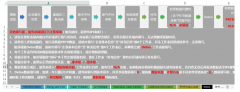
FDA510K评审周期
Day 1:FDA receives 510(K) submission
↓
By Day 7
FDA sends Acknowledgement Letter.
OR
FDA sends Hold Letter if unresolved issues with User Fee and/or eCpoy.
↓
By Day 15:
FDA conducts Acceptance Review.
FDA informs submitter if 510(K) is accepted for Substantive Review or placed on RTA Hold.
↓
By Day 60:
FDA conducts Substantive Review.
FDA **municates via a Substantive Interaction to inform the sunmitter that the FDA will either proceed with Ineractive Review or that 510(K) will be placed on hold and Additional Information is required.
↓
By Day 90:
FDA sends final MDUFA Decision on 510(K).
↓
By Day 100:
If MDUFA Decision is not reached by Day 100,FDA Provides Missed MDUFA Decision Communication that identifines outstanding review issues.
联系我时,请说是在得易搜分类信息网看到的,谢谢!









 未上传身份证认证
未上传身份证认证  未上传营业执照认证
未上传营业执照认证